

-
-
-
Application
-
-
Essential Guide to Medical Electronic Device Design
2023-04-04
The medical electronic device industry grew significantly over the last few decades following the exponential advancements in electronics, computation, and life sciences. Also, the rise of personal and mobile healthcare increased the demand for small, wearable, and low-power technologies, adding heat to the medical devices market. Therefore, it is not surprising that more and more electronics developers are entering the field. But designing medical electronics is not an easy task, and the developer must be prepared to face challenges and regulations that are not required in consumer electronics.
One of the most common mistakes is not considering the relevant regulations early in the design process, leading to expensive modifications or even the complete redesign of the device. Therefore, it is important to know the regulations and best practices before starting a new project. In this article, we will discuss the most important regulations and standards for medical electronic devices, focusing on electronic systems.

Tests & Standards for Medical Electronics
The regulation for medical devices depends on the country of origin: for instance, the Food and Drug Administration (FDA) is the main regulatory agency in the US, whereas the European Medicines Agency (EMA) is the EU equivalent. Nevertheless, design guidelines are typically based on the International Electrotechnical Commission (IEC) standards, in particular the IEC-60601-1 and IEC-60601-1-2. By adhering to these standards, the product becomes “601 compliant” which is crucial for certification approval and market release.
The IEC-60601-1 defines medical electrical equipment as any “electrical equipment having an applied part or transferring energy to or from the patient or detecting such energy to or from the patient”. In other words, a device that expends a period in contact with the user and/or exchanges energy with the body. Some examples of medical devices are ECG, EEG and EMG sensors, ultrasound imaging, heart rate monitors, glucometers, digital thermometers, and pacemakers. These devices must pass several EMI/EMC, ESD, and power supply immunity tests for certification, maintaining both safety and essential performance during and after the tests.
EMI/EMC Tests
These tests ensure that the medical device is compatible with the electromagnetic environment, that is, if the device is not a noise generator and can withstand the background EMI without problems. In terms of emission, the device must comply with the International Special Committee on Radio Interference (CISPR), in particular the CISPR 11 standard. There are two types of EMI: the conducted and the radiated.
The conducted EMI refers to interference energy transmitted through cables, whereas the radiated EMI corresponds to wireless interference. In the CISPR 11, the frequency range for tests should cover 150 kHz – 30 MHz (conducted) and 150 kHz – 18 GHz (radiated), with maximum amplitudes depending on the device class. Moreover, the conducted power line harmonics must be filtered out in accordance with the IEC 61000-3-2, especially if the device uses switched-mode power supplies. Conducted noise can be filtered using ferrite beads, line filters, and chokes. Radiated EMI regulation may change from country to country, depending on the regulatory agency.
Radiofrequency susceptibility is more critical in medical devices, as they must be robust enough to continue to operate even under problematic EMI conditions. The tests are specified by the IEC 60601-1-2, with a frequency range of 80 – 2700 MHz for radiated immunity and 0.15 – 80 MHz for conducted immunity. The maximum amplitudes vary with the device, which can be classified as home healthcare equipment or professional medical facility equipment.
During the test, the device is submitted to several levels of RF interference in an anechoic chamber, and any functionality failures during and after the test are analyzed and reported. The device should also be immune to low-frequency magnetic interferences (50/60 Hz) induced by the power lines (IEC 61000-4-8).
To pass the EMC tests, be careful with the circuit and PCB design, to prevent high impedance nodes, loops, and other parasitic antennas. The proper implementation of filters, common-mode compensators, shields, and robust signaling techniques (such as differential lines and coaxial cables) can significantly improve EMI immunity.
Power Supply Immunity Tests
Medical electronic devices connected to the power distribution system cannot be too susceptible to power supply instability, as it would put the patient at significant risk. Therefore, the IEC-60601 also establishes several immunity tests, covering fast transient surges, flickering, and power instability. The IEC 61000-3-3 standard describes power fluctuation and flicker testing, specifying the maximum amplitudes and periods of voltage variation that the device must endure during the inspection. For voltage dips testing, the IEC-61000-4-11 is used.
Medical devices must operate correctly during and after voltage dips of 95% for up to 5 seconds, which can be accomplished using batteries or capacitor banks. Finally, the IEC-61000-4-4 and IEC-61000-4-5 are used to assess the device’s susceptibility to bursts and surges, with different lengths and amplitudes (up to ±2 kV).
Electrostatic Discharge Tests
Medical electronics, especially small circuits with direct contact with the user, are susceptible to ESD, so robust ESD protection is a must. The standard for the ESD protection testing is based on the IEC-1000-4-2 standard. During the assessment, the device under test is submitted to ±8 kV in contact discharges and ±15 kV in air discharges. The device must present error-free operation during this test, so basic ESD immunity is fundamental in any design, but are especially important when high voltages are used, such as in defibrillators.
Risk Management Matrix
A powerful tool for medical system design is the risk management matrix. It is used to provide quantification metrics for each possible risk associated with the medical device during any operation mode and possible fault. The international guidelines for the matrix are defined by the ISO 14971:2019 standard, which also includes medical software and in-vitro devices. It addresses risks related to biocompatibility, electrical harms, radiation, and usability among others.
A risk management matrix can be designed created by distributing the failure likelihood (rare, unlikely, likely, expected, and certain) in one axis, and the consequences of the failure (minor, moderate, serious, major, and catastrophic) in the other axis, and filling the cells with the level of acceptance. One example is shown in Table 1.
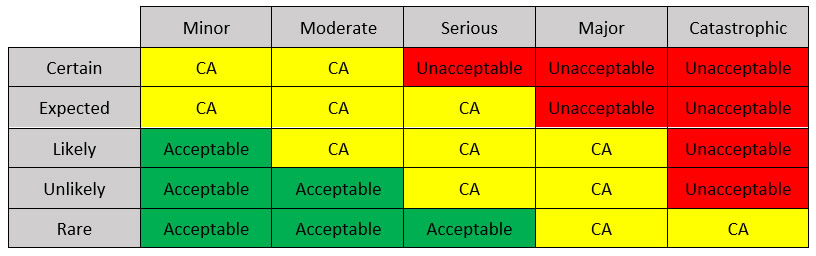
Table 1: A possible Risk Management Matrix (CA: conditionally acceptable).
In the example matrix, there are three zones: the acceptable region, the conditionally acceptable (CA) region, and the unacceptable region. Each corresponds to a combination between failure probability and consequence. The consequences are related to specific real-life outcomes depending on the device and application, including system loss, major system damage, minor patient injury, severe patient injury, and patient death.
Risks that fall in the acceptable region require minor modifications or no modification at all, while risks in the CA region require the identification of mitigation mechanisms to become acceptable. Unacceptable risks must be eliminated.
Patient Isolation
To ensure that the problems related to the electronic modules do not affect the patient’s body, it is crucial to provide adequate isolation between the circuit and the user. It is one of the most important parts of medical design, and inappropriate isolation is one of the main reasons for rejection.
The IEC-60601-1-1 states that no electrical DC path can exist between the user and the device, as it can cause local burns and harm. Therefore, any signal reading or injection must occur via decoupling capacitors or wirelessly. The standard describes well-defined rules for leakage current, voltage amplitude, energy, and signal frequency applied to the patient, as well as the dielectric isolation and clearance necessary depending on the voltage levels. Also, any part not connected to the patient must provide good levels of insulation, which can be done by proper clearance and coating.
Isolation transformers, optocouplers, high-pass filters, and decoupling capacitors are some of the tools that can increase safety and ensure that the design passes the certification process. Moreover, any external power supply, regulator, or converter implemented must comply with the IEC-60601-1 standard.
Laboratory Equipment
There are many devices used in medical practice and research that need no contact with the patient at all, which are considered laboratory equipment. In these cases, the device must be designed in accordance with the IEC-61010 standard, which establishes the safety requirements for laboratory electrical instruments and equipment, and the IEC-60610-1 guidelines. Devices such as microscopes, autoclaves, chemistry analyzers, cytology equipment, and cell culture incubators are examples of laboratory medical equipment.

Medical Electronics Certification Path
An integral part of the medical design loop is the certification process, which decides whether your device is ready to market or not. This process is defined, controlled, and executed by a regulatory agency, varying from country to country. For instance, medical devices are regulated by the FDA in the US, by the EMA in Europe, and Health Canada in Canada. In this article, we are going to focus on the FDA regulation, as the US medical market is huge, and the FDA complies with most regulations around the world.
Device Classes
The FDA rules for product certification depend on the nature of the medical device. In this sense, the agency defines three different device classes to specify what type of process the product must undergo, including premarketing application, approval, and controls. Medical devices are classified according to the risk level:
- Class I: devices that are not intended for life-support and/or life sustaining applications, and do not represent a significant risk to the user in case of any failure. The certification process is much more relaxed for Class I devices, as most of them can be released to the market without any pre-market notification or approval. However, every Class I device must comply with the general FDA controls. Class I covers the majority of medical devices in the market, including bandages, stethoscopes, and surgical masks. Although most Class I devices do not contain critical electronic parts, electric toothbrushes and hospital beds are included in the class.
- Class II: these devices pose a moderate risk to the patient/user, as they typically require long periods of contact with the body and often have invasive parts. Different from most Class I devices, Class II equipment needs to pass through the pre-market notification process, also called 510(k). Some devices, however, are exempt from the 510(k). The FDA also defines special controls to regulate Class II, which vary with the application. Some examples of Class II devices are electrical wheelchairs, x-ray machines, infusion pumps, and contact lenses.
- Class III: this class covers high-risk medical devices, including equipment for life-support and sustain, implants, and any equipment that presents a significant risk of injury or illness to the patient/user. Approximately 10% of the approved medical devices are Class III, including pacemakers, defibrillators, cochlear implants, and high-frequency ventilators. General and special controls are not enough to regulate Class III devices, and the majority need to pass through the premarket approval process (PMA). Exempt Class III devices still need to fill the 510(k) notification but are free from the PMA.
Market Release
FDA defines several steps and standards to bring a medical device to the market, including general and specific controls, pre-market notification – 510(k), and premarket approval (PMA). The certification path depends on the class and nature of the device, as well as the exemptions applied. Therefore, the first step consists in identifying the class of the device, which can be done through the classification panels system developed by the FDA.
Classification Panels
One of the easiest ways of identifying the class of a device is by finding a matching description in the classification panels. The FDA divided more than 1700 types of medical devices into 16 different panels (or categories). Each panel is associated with a medical specialty, including “General Hospital”, “Microbiology”, and “Orthopedic”, which are then divided into device types. For instance, the “Orthopedic” specialty covers more than a hundred types, such as “Bone Cap” and “Intramedullary Fixation Rod”. Each item contains a brief description and the corresponding class (I, II, or III) of the device. The classification panels can be accessed in the FDA website.
General and Special Controls
The FDA also establishes a basic set of design rules that are common to all medical devices: the general controls. It regulates, among other items, device adulteration, misbranding, registration, premarket notification, and restricted devices. It also provides general guidelines for best manufacturing practices. In the case of exempt Class I devices, compliance with the general controls is the only requirement before market release. However, for non-exempt Class I devices and most Class II devices, the General Controls are not enough, so the FDA defines the Special Controls. Post-market surveillance, premarket data requirements, special labeling requirements and patient registries are some of the topics included in the Special Controls section.
Pre-market Notification 510(k)
In addition to the Special Controls, Class II devices require pre-market notification submission for FDA approval. This process is often called 510(k), which corresponds to the premarket notification section in the Food, Drug, and Cosmetic Act (FD&C). The 510(k) submission is a thorough description of the medical device, including technical and functional information concerning effectiveness, safety, and operation modes. For approval, the FDA verifies if the device is “substantially equivalent” to another medical device already in the market. Therefore, the 510(k) process is basically a certification by equivalency. Most Class II and some exempt Class III devices only need to submit a 510(k) notification before market release, so searching for equivalent equipment to avoid the need for a PMA is a fundamental step in these classes.
Pre-market Approval (PMA)
Finally, the last step in the FDA certification process is the Pre-Market Approval (PMA). The PMA process includes a series of clinical trials and laboratory experiments to certify the effectiveness and safety of the medical device through sound and consistent data. Therefore, it is an expensive, laborious, and time-consuming process, and failing the PMA can be disastrous to the company. The FDA provides well-defined guidelines and standards for every test, which can be consulted by the engineer during the design process to prevent recalls. Most Class III devices are considered elevated risk equipment, and cannot be marketed legally without a PMA certification, so it is important to identify the class of the device and integrate the PMA requirements early on in the design flow.
Revving Up the Future: The Importance of Automotive Semiconductor Chip in the Connected Car Era
Automotive semiconductors are a major component used in numerous advance driver assistance systems (ADAS) technologies, which helps ADAS systems perform efficiently and can detect and classify objects on the path of vehicle, and accordingly alert the driver about the nearby surroundings and road conditions
04-25
2023
Essential Guide to Medical Electronic Device Design
The medical electronic device industry grew significantly over the last few decades following the exponential advancements in electronics, computation, and life sciences. Also, the rise of personal and mobile healthcare increased the demand for small, wearable, and low-power technologies, adding heat to the medical devices market. Therefore, it is not surprising that more and more electronics developers are entering the field. But designing medical electronics is not an easy task, and the developer must be prepared to face challenges and regulations that are not required in consumer electronics.
04-04
2023
Electrical Components for Medical Devices
WeSun Electronic Technology manufactures superior quality electrical components for utilization in medical devices. All technology in medical environments must meet strict standards for radio frequency interference, electrical noise emission and susceptibility standards. All electrical components manufactured by WeSun Electronic Technology are tested and guaranteed to meet all FCC and European standards for electronic components used in medical equipment. From RFI filters to barrier terminal blocks, WESUN is the leading source for electrical components manufactured for medical technology utilization.
04-04
2023
Xi'an Wesun Electronic Technology Co.,Ltd.
GET A QUICK QUOTE & DETAIL INFORMATION
PRODUCTS LIST
CONTACT US
Address: Greenland SOHO Building B 1509, Zhangba 1st road,High-tech Zone Xi’an,Shaanxi 710075 China.
Email: sd02@wesunele.com
TEL: +86-029-81120758
Mob: +86-132 9910 7326
Copyright © 2023 Xi'an WeSun Electronic Technology Co.,Ltd.